Kicking off the webinar, Dr. Frederick Ch'en, partner in the Hogan Lovells Intellectual Property, Media, and Technology practice, APAC lead for Life Sciences, and Office Managing Partner of the firm’s Tokyo office, noted that whether or not your organization (or a competitor) has a product that has been selected for negotiation under the IRA, the impact of the legislation looms over the industry. Manufacturers of a product having a payer mix with an expected spend of at least U.S. $200 million in annual Medicare sales must now grapple with the impact that the IRA will have on launches and the partnerships often required to bring an asset to market. That includes a new focus on launch timing, from managing the timing of an New Drug Application (NDA) or Biologics License Application (BLA)’s submission to maximize the IRA time clock, to ensuring a company is fully launch-ready as of “day 1” following a product’s first FDA approval with a value story that is fully developed.
New price controls will have a profound impact on U.S. drug pricing
Alice Valder Curran, partner in the Hogan Lovells Health practice, explained that the IRA includes major provisions relevant for drug pricing and reimbursement, including an entirely new Drug Price Negotiation Program, which seeks to lower the prices of certain Medicare high spend drugs by imposing a maximum fair price (MFP) on specified qualifying single source drugs (QSSD). Under the program, certain NDA drugs at least seven years post-approval and certain BLA biologics at least eleven years post-approval and for which there is no generic / biosimilar on the market are subject to “negotiation” and application of an MFP by year nine and 13 post-approval, respectively. Our team has previously outlined key details of these provisions here and here.
Ms. Valder Curran highlighted that manufacturers of a product having a payer mix with an expected spend of at least U.S. $ 200 million in annual Medicare sales must grapple with the impact that the IRA will have on their U.S. product launches. She also framed a number of considerations that may impact a manufacturer’s litigation strategy and possible licensing opportunities, as also discussed in more detail below.
When it comes to the interplay between innovator and prospective generic / biosimilar launches, Ms. Valder Curran pointed out that a generic / biosimilar must be marketed before the end of the “negotiation” period in order to avoid application of MFP, with later entry creating other potential unique dis/incentives for generic entrants and their branded counterparts. Ms. Valder Curran discussed the factors underlying the timeline depicted below, which shows the selection and negotiation dates for initial price applicability year (IPAY) 2026, and highlights the impact of a generic / biosimilar launch, respectively:
- before the QSSD selection date;
- before the end of the negotiation period;
- after the end of the negotiation period but through the first three months of IPAY 2026; or
- on or after April 1 of IPAY.
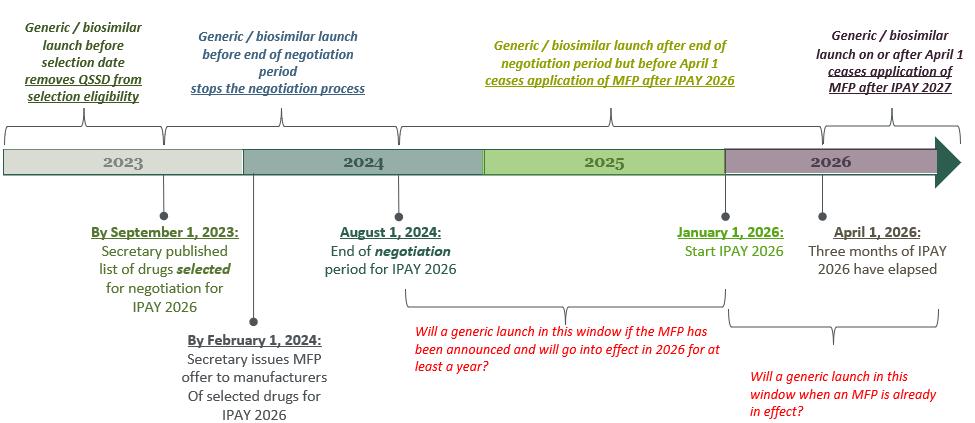
Industry stakeholders should be on the lookout in the coming months leading into the November U.S. presidential election to understand where the MFP for each of the first ten “negotiated” drugs will land relative to their respective wholesale acquisition cost (WAC).
Evaluating and addressing exclusivity loss
Howard W. Levine, partner in the Hogan Lovells Intellectual Property, Media, and Technology practice, began his analysis by discussing the historic incentive structure created by the Hatch-Waxman Act, including its intended balance between generic and brand drug companies in the U.S. market. The IRA has further shifted the incentive structure, he noted, because patent and regulatory exclusivities will not protect from an MFP. He also discussed how the IRA places potentially greater emphasis on each of:
- a BLA vs. an NDA pathway, where possible;
- protecting backup molecules in addition to a lead compound; and
- alternative IP protections, such as know-how and trade secrets.
Mr. Levine then discussed how companies should consider assessing the impact of the IRA’s new “MFP cliff” as compared to the well-known “patent cliff” when evaluating possible litigation scenarios. He offered a number of factors (including timelines for protection, revenue impact of price controls vs. generic / biosimilar entry, protecting alternative compounds vs. multiple uses) that companies should evaluate for each of their product(s) / portfolio(s). He then discussed the impact of the generic and biosimilar launch timing considerations framed by Ms. Valder Curran.
Finally, Mr. Levine noted that settlements and / or agreements may ultimately take a larger role in informing litigation scenario planning, but cautioned that companies will also need to consider the implication of possible antitrust issues on any of their analyses.
Transactions and collaborations in a post-IRA regime
Cullen G. Taylor, partner in the Hogan Lovells Intellectual Property, Media, and Technology practice and head of the Life Sciences Licensing and Commercial Transactions practice, opined that pharmaceutical and biotech companies may want to think differently about their licensing and agreement relationships in a post-IRA world. First, he presented a framework that companies could use in evaluating their existing and future product(s) / portfolio(s) for MFP risk, based on factors that may impact selection of products for “negotiation” and application of an MFP, as well as the projected initial IPAY for a product in a particular indication. He also discussed the potential impact of timing where approvals across multiple indications are anticipated for a particular asset.
He then explored whether out-licensing opportunities might provide opportunities to mitigate these risks across molecule(s), especially where a therapy may offer patient benefits across multiple possible indication(s). He cautioned, however, that the costs and benefits of these strategies must be weighed carefully, given the challenges inherent in indication-specific out-licensing.
Mr. Taylor outlined a number of new IRA-specific risk considerations, and mitigation strategies, from both licensee and licensor perspectives. For example, post-IRA, licensees may have higher interest in retaining rights to backup compounds and combination therapies, as well as increased need for flexibility in sublicensing and launch strategies, and a desire to ensure the diligence and economic provisions account for the impact of the MFP. Licensors will be focused on ensuring they are appropriately compensated for granting rights to backups and combination therapies, as well as ensuring a licensee carries out a timely launch. Our team has also described the impacts of the IRA on licenses and collaborations here. Ultimately, stakeholders will need to grapple with the potentially negative impact that the IRA will have on the economics of a drug asset through mitigating and appropriately allocating risk under the new framework.
The full webinar is available here and you can view the slides here.
